

Pfizer's own data showed vaccine creates net loss on hospitalisations
How did regulators miss this?

When the regulators made their decision to put the mRNA vaccines onto the market, they did so based on evidence given to them by the vaccine manufacturers themselves. In the UK, the regulators called this process a ‘rolling review’, and after it they were determined to be safe and effective enough for emergency use. We still hear commentators use this ‘safe and effective’ phrase, and we can fairly presume they’re referring to the data used by the regulators to get these products on the market.
So what does that data actually show?
To illuminate this I’ll refer to three documents from which we can get broad agreement. Focusing on the Pfizer vaccine, I’ll be referring to the ‘Public Assessment Report’ from the MHRA, the UK regulator. The report references the same data detailed in the ‘FDA’s briefing document’ dated December 10th 2020. I’ll also reference the first published study about the Pfizer vaccine published in the New England Journal of Medicine.
To show they had some handle on safety, in their report, the MHRA showed the rates of serious adverse events “temporally associated with the vaccine.” They defined these events as “any untoward medical occurrence… that resulted in death, was life-threatening, required inpatient hospitalisation, resulted in persistent disability/incapacity or was a congenital abnormality/birth defect.” To keep things simple, you can imagine a serious adverse event as a medical occurrence that landed the patient in hospital, I’m going to refer to them as SAEs. These SAEs may or may not have been associated with the vaccine.
However, the investigators gave us some steer on this issue of causation. According to Table 12, some of the SAEs were “related to the investigational product”. Of the 21,621 eligible participants in the vaccine group, four of them had a related SAE following vaccination, whilst in the placebo group, there were zero. This imbalance in outcomes works out to 1 in 5,405 vaccinated people having a related SAE.
The report also mentions a total of 126 SAEs vs 111 in the placebo. Whether they are related or not is for the investigator or the regulator to determine. However, in a strange editorial decision, commenting on these figures the MHRA said “there was one serious adverse event of anaphylaxis 9 days after Dose 2 of BNT162b2; this was due to a bee sting. No significant concerns are raised.” The bee sting detail, intentionally or otherwise, seems perfectly placed to dampen the imbalance in SAEs between the groups. It’s the only real comment we get from the MHRA on these numbers, and as such, it characterises the 126 SAEs as not really worthy of attention. And yet right there in Table 12, the investigator says 4 of the SAEs were considered related to the treatment. There is no mention of this in the body of the MHRA report. Why?

The omission might be somewhat explained by the MHRA’s new strategy in which they have moved “from watchdog, to enabler.” Dr June Raine, the head of the regulator, stood before an applauding Somerville Oxford College and proudly declared her vision. Is that what the MHRA was doing here? Instead of closely interrogating the risk for this novel therapy to do harm, they instead glossed over the inconvenient safety data to fulfil their role as ‘enabler’? With our regulator proudly disinterested in being a watchdog, allow me, however imperfectly, to take a critical look at the number the MHRA made no mention of. But first - please subscribe and share my substack if you haven’t already!
Four SAEs in the vaccine group, vs zero in the placebo, suggests that 1 in every 5,405 people would have a severe adverse event related to the treatment. It’s actually a fairly large number. That event, according to the MHRA’s own definition, would put the patient in hospital. As such, clearly understanding the rate at which the vaccine was creating hospitalisations is key to understanding the risk/benefit of the product, something that in normal times we would expect our regulator to be very keenly interested in. To get the risk / benefit, we need to compare like for like, so we need to know how many people the vaccines might keep out of the hospital.
It’s a piece of data that the MHRA were aware of, but again, for some reason they chose not to include it in their public assessment report. A “key secondary objective was to evaluate the efficacy against confirmed severe COVID-19” they said on page 27, but I found no reference to the data in their public report. The FDA detailed it, and it was also detailed in the New England Journal of Medicine Study published on December 10th 2020. This interesting piece of data was of course buried on page 11 of the appendix. So let's take a look.
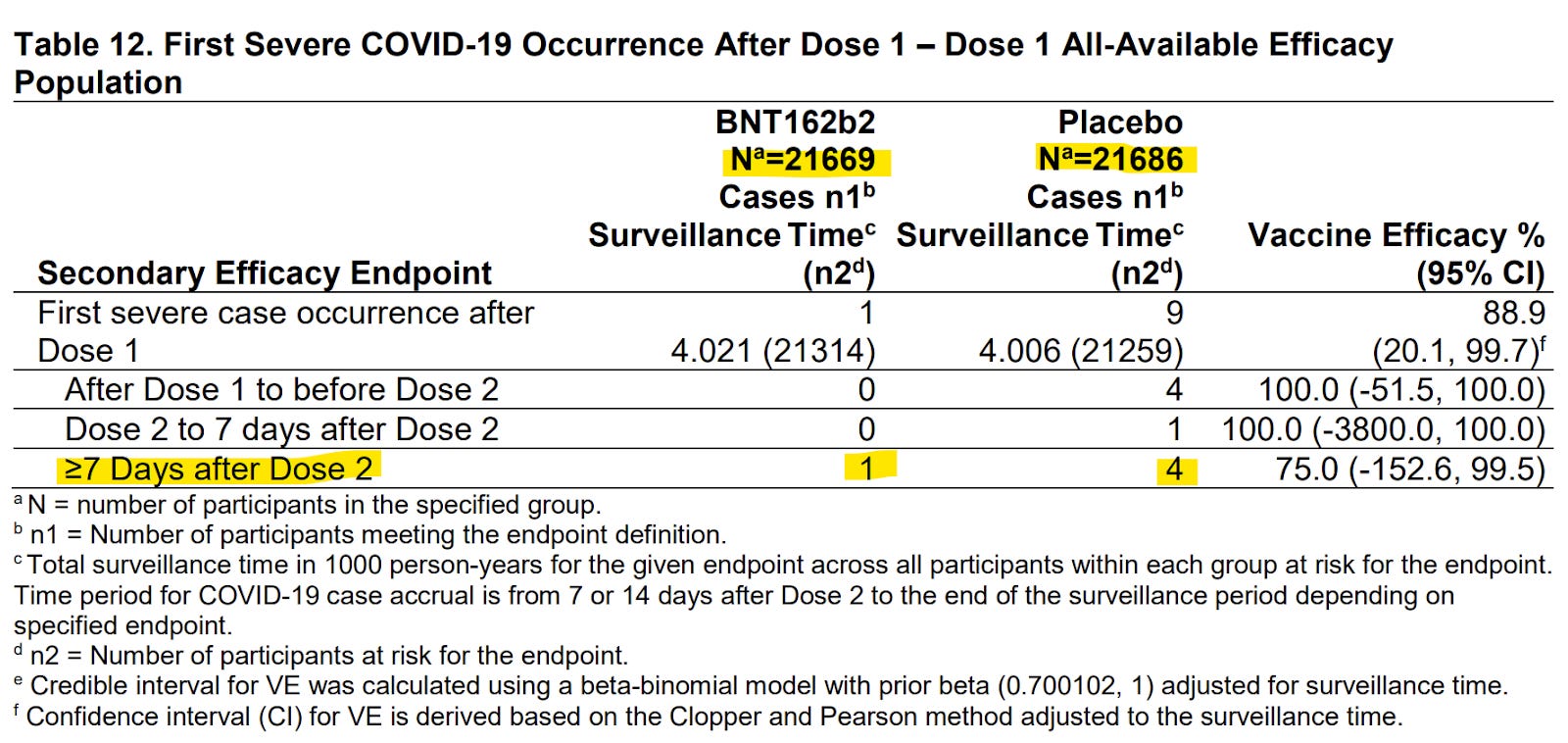
Above is the data we need. It compares the rate at which patients in each group were contracting serious Covid-19 within the study period. Serious Covid-19 had a technical definition, but essentially, it would be enough to land you in hospital. We can use the data in Table 12 to calculate a figure called the ‘Number Needed to Treat’, which means exactly what it sounds like. It’s the number of patients we need to treat in order to prevent one of those outcomes in the real world, in this case, a “severe Covid-19 occurrence”. We almost never hear pharmaceutical companies present their data this way. To answer why would be to speculate, but perhaps it’s because the ‘NNT’ makes it difficult to hide low efficacy. It's also a very intuitive way for the public to understand how well a drug works. But the thing that stands out most to me, is that baked into the ‘number needed to treat’ is the actual risk of a patient becoming ill with the condition in the first place.
Because we have two groups to compare, the vaccine vs the placebo, we can see by how much the vaccine reduced the risk of hospitalisation within a given period. We can then calculate how many people we would need to treat in order to prevent one of those outcomes out in the real world. In the example below, we’re assuming two doses as the treatment.

It tells us that we need to fully vaccinate 7,230 people in order to prevent one serious COVID-19 outcome. But using the same dataset, we can see that serious adverse events related to the vaccine were running at 1 in every 5,405 people. Can you see the problem? The data suggests a net loss in hospitalisations. For every person we keep out of hospital for Covid-19, we’re putting 1.3 people into hospital with serious adverse events related to the vaccine. Obviously Pfizer isn't going to present this fact explicitly to the watchdog, but any watchdog focused on regulating rather than “enabling” would make these calculations and ask very tough questions of the manufacturer. Why wasn’t this spotted?
Even with these figures, I think we’re being generous to Pfizer. If I were to put my ‘regulator’ hat on, I’d start from the perspective that the drug doesn’t work, it isn’t safe, and Pfizer is trying to pull the wool over my eyes. Why? Because Pfizer have been sued for “causing false claims to be submitted to government healthcare programs” in the past. People died as a result of their chicanery, which resulted in the biggest legal settlement in US history. Being in this sceptical mindset, I wouldn’t be moved by the number of SAEs that the investigator considered related to the vaccine, I’d want to look for evidence of a problem myself. I’d start by looking at that total difference in SAEs between the vaccine group and the placebo group. Since the two groups were properly balanced by age, comorbidities and demographics, and they were a good size, we can compare any differences in outcome. All things being equal, the outcomes in SAEs should be somewhat equal too. This is one of the major attractions of a controlled trial - balanced cohorts.
With 126 serious adverse events in the vaccine group and 111 in the placebo - that’s an imbalance of 15. Arguably, we can therefore say the vaccine caused 15 serious adverse events per 21,669 people. That’s one in every 1,444 people.
There’s very little detail in the regulators reports on this discrepancy. We only get qualitative statements suggesting “No significant concerns are raised.” The assumption, both from the FDA and the MHRA, was that the events being picked up “occurred no more frequently than expected”. What they’re arguing is that we’d expect to see some serious adverse events anyway, and any difference between the two groups isn’t large enough to worry about. But if that was the case, we’d expect to see a random ‘background’ distribution of SAEs over time. We wouldn’t expect to see them clustering around when the vaccines were administered, that would be a ‘temporal association’. It’s something we could test for using Pfizer’s data, and yet, neither the FDA, the MHRA, nor the study authors make any effort to look for this pattern. The MHRA spent nine pages investigating the expected adverse events like sore arms, and just four paragraphs on the serious adverse events. The pattern was identical in the FDA report. Why would the regulators be less interested in the adverse reactions that could kill people? Why didn’t they properly interrogate this data?
I wanted to check for an association, so I spent a long time cleaning up the dreadfully formatted data which Pfizer was forced to release to the public. Using this pdf - yes Pfizer released the data as PDFs - I was able to extract the case notes on the serious adverse events. It’s on page 2471 onwards if anyone is interested in checking it. After cleaning it up, I was able to plot out the count of SAEs by week number. The graph below shows us the number of SAEs in each group, by the week number following any dose of vaccine (dose 1 or dose 2). There’s two things that stand out immediately: firstly, the SAEs are ‘temporally associated’ to the administration of the doses, because they clearly decline in number over time. We see an ‘expected’ rate (as the FDA put it) of less than 10 serious adverse events per week on the tail of the graph at 12 weeks out. In the weeks closest to the administration of a vaccine, there are 300% more SAEs than the baseline. It’s quite clear they’re associated with the injections.

Secondly, in the first two weeks following a vaccination, the placebo group stands out as…strange. Why are we seeing more SAEs in the placebo group over the live vaccine? It’s not what you would expect to see, because the placebo patients are being injected with an inert saline solution. By weeks 3 to 5, the rates drop right back to what you might reasonably expect, fewer SAEs in the placebo group, eventually tailing off to a low baseline of around 5 events per week. So what’s going on here?
Pausing the article here, to ask, if you’re reading this and like the work I’m doing, and believe me it’s a lot of work, I’d really appreciate a subsciption to the digger. If you or someone you know would like a subscription, there’s even ways to gift it to people. Thanks.
Being mindful of Pfizer's habit of submitting ‘false claims’ to regulators - the data certainly has the appearance of a potential fudge. Were investigators under pressure to report additional SAEs into the placebo group across the first two weeks? Doing so hides the uptick of side effects in the vaccine group within 14 days of receiving a shot, and this is the period the MHRA and the FDA were both most focused on. If you wanted to hide a safety signal, your distribution of SAEs might look quite similar to the graph above.
For such a thing to be possible, the trial staff would have to know which patient is on which treatments. They’re not supposed to know this information in a randomised control trial, so could such a thing happen? The Brook Jackson whistleblower case, reported in the British Medical Journal, tells us this is exactly what happened on the trial. Staff and patients did become unblinded during the trial. The patients had the potential to know which treatment they had, and staff had the same. Brook Jackson even told me that investigators were forging documents and changing the diagnosis of patients. This is exactly what you’d need to do if you wanted to nudge the data in your favour. If the staff know which patient is receiving the placebo, they over-report SAEs to that group to hide a commercially damaging safety signal. Map this onto Brook Jackson’s observation of trial staff altering patient’s diagnosis, and it doesn’t look good. The commercial motivation is there, there’s a clear history of pulling the same tricks, the trial staff were unblinded and in a position to do it, and a whistleblower tells us that such alteration of patient data was going on.
Perhaps there’s an explanation for the pattern that doesn’t require a fudge. Perhaps. But as regulator, I’d have very little of the good will required to give the benefit of the doubt.
Perhaps the investigators got lucky? The average rate of SAEs across the whole period was 10 per week, and if we exclude those the first two weeks, the average was just 6. Looking at it this way suggests there was an unexpected ‘triple bump’ in SAEs in the placebo group. Through folly, farce or fortune, that jump in SAEs hid a potential safety signal. It may well have hidden a stop signal.
Were there qualitative differences in the kinds of SAEs being reported to each group? If my theory is correct, we might be able to see evidence of it by looking at the kinds of SAEs reported. Might we see more ‘soft’ reports? As a sanity check, I extracted the case notes for the SAEs across the first 14 days of the trial. I cleaned them up, separated them into placebo and vaccine groups, and ran them through a word cloud generator to give a quick steer as to the kinds of events being reported. I’m posting the results below, and I’ll leave it to you to work out which set is which. Can you tell?


So what can we make of all this? Even if we don’t accommodate the ‘triple bump’ in SAEs we discovered, the data still shows 15 additional serious adverse events in the vaccine group. That works out to 1 in every 1,441 people. When we add the figure to our risk benefit calculation, it moves dramatically in the wrong direction. To keep one patient out of hospital, we need to double vaccinate 7,230 people, and of those people we’d expect to see five seriously injured from the vaccine.
1 patient prevented from hospital, 5 patients put into hospitalised.
Rather than having “no significant concerns” as the MHRA put it, or “no clear basis to suspect vaccine-related risk” as the FDA said, I’m inclined to disagree. There was a safety signal sitting right inside the data submitted to the regulators. All they had to do was look for it. Regulators on both sides of the pond dropped the ball. Even at a surface level, the imbalance in SAEs between the vaccine and placebo group is worth paying attention to, but they gave it no attention at all. Had the regulator plotted out the timings of those SAEs, they’d have seen a temporal association with vaccine administration. It’s not clear they looked any further than the tables provided to them by Pfizer. Whether a trial error or an anomaly, the distribution of SAEs in the placebo group warped the results in the favour of the manufacturer, and this data was easily spotted if the regulators had bothered to look. Making corrections for it may worsen the safety profile of the vaccine even further.
With neither regulator bothering to interrogate the safety signal, what followed was an indiscriminate vaccination campaign with a novel product for which the data showed a net loss on hospitalisations. Around 18 months after the rollout, federal German health authorities released data confirming the problem. The vaccines are associated with a 1 in 5000 chance per shot of a serious adverse event. So for the double vaccinated, that number is 1/2500. For the boosted it’s even higher. If mainstream commentators have reacted at all, they’ve reacted with surprise to these figures from the German government, sometimes with a hint of denial. But the rate the German health agency revealed is actually lower than the rate you can find in Pfizer’s own data if you’re reading it critically. Given their track record, I don’t know how anyone can read their data any other way.
Epilogue
Answering some expected criticism:
I expect my critics to say that the difference of 15 between vaccine and placebo isn’t large enough to draw conclusions from. It’s an argument relied on to some extent by the regulators themselves. In which case, which data do you suggest we use to determine the product is safe? This was the study. If the 43,000 sample size isn’t large enough to draw conclusions from, then what is? How large does the safety signal have to be before a study of 43,000 can satisfactorily detect it? This criticism implicitly acknowledges that the study was incapable of detecting a safety signal affecting as many as 1 in 1440 people, which is a disaster. In such a scenario, we cannot use the study to claim the product is safe at all.
To add to that, what if there was an imbalance of 30 between the vaccine and placebo group? We’d then be looking at a safety signal affecting 1 in every 722 people. Are we suggesting that the study is incapable of picking up safety signals until they start happening at rates higher than 1 in 1400 or 1 in 722 people?
Finally, doctors and patients have to know the side effect profile of these novel products for informed consent to be realised. Using data available at the time these products were rolled out, can you calculate the rate of expected side effects so that we can have a reliable risk / benefit calculation? If so, what was the rate of serious adverse events to be expected?
This is an open thread so please… feel free to
Brilliant analysis! Everyone involved in this debacle deserves hard time.
one assumption we're making is that the placebo group actually got saline. What if they didn't? What if they got lipid nanoparticles without the mRNA? What if they were injected with everything that's in the "vaccine" except the mRNA? At this point there is evidence that the lipid nanoparticles themselves are toxic even without the mRNA. If the placebo group did not get saline and instead got the vaccine concoction sans mRNA, then that would explain the serious reactions in the placebo group in the first couple of weeks.